In the paper “Grisoni, F., Consonni, V., & Vighi, M. (2018). Acceptable-by-design QSARs to predict the dietary biomagnification of organic chemicals in fish. Integrated Environmental Assessment and Management, 15(1), 51–63. https://doi.org/10.1002/ieam.4106“, the authors presented some regression models to predict the laboratory-based fish Biomagnification Factor (BMF) of chemicals.
Originally we produced a bespoke software, named BMFpred, to replicate the models and results of this paper. BMFpred was phased out in favor of a more generic solution to create and deploy QSAR/QSPR models based on our software tools alvaModel and alvaRunner. In particular, the BMF models can now be applied to your molecules using the alvaRunner project we present here.
Additional information about the original paper is available at the In silico Biomagnification Factor prediction project.
alvaRunner project
This alvaRunner project contains three models:
- a KNN regression model, specifically a weighted Nearest Neighbour Regression model
- an ordinary least squares (OLS) model
- a Consensus model defined as the arithmetic mean of the values predicted by KNN and OLS models
The KNN model includes four descriptors:
- MLOGP2: the square of the Moriguchi octanol-water partitioning coefficient
- nBt: the total number of bonds
- B02[N-O]: the presence/absence of a nitrogen atom and an oxygen atom separated by two bonds
- F06[C-C]: the counter of carbon pairs separated by six bonds
The OLS model is comprised of seven molecular descriptors. As well as the KNN model it includes the MLOGP2 and B02[N-O] descriptors. Additionally, it includes X0Av, X1Per, SaaaC, VE1_B(m) and B03[N-Cl] molecular descriptors.
Grisoni highlighted that X0Av (average connectivity index of order 0) is related to the fraction of atoms with many valence electrons and to unsaturated/aromatic bonds. Additionally, X1Per (perturbation connectivity index) is considered sensitive to the presence of heteroatoms, molecular shape and presence of multiple bonds. SaaaC is an atom-type electrotopological state index, it is related to the electron accessibility of specific-atom types, specifically SaaaC is the sum of the E-states of Carbon atom connected to 3 aromatic atoms. The authors also stated that on the considered dataset, VE1_B(M) (coefficient sum of the last eigenvector (absolute values) from Burden matrix weighted by mass) is related to the molecular size, the branching, the number of multiple bonds and the number of cycles. Finally, B03[N-Cl] similarly to B02[N-O] indicates the presence/absence of a nitrogen atom and a chlorine atom separated by three bonds.
In the following table we present the scores of the three models of the alvaRunner project:
| Model name | Training | Test | ||||
|---|---|---|---|---|---|---|
| R2 | Q2CV | RMSE | RMSECV | R2 | RMSE | |
| M1_KNN (k: 6, Manhattan) | 0.747 | 0.756 | 0.536 | 0.527 | 0.777 | 0.544 |
| M2_OLS | 0.758 | 0.739 | 0.525 | 0.545 | 0.757 | 0.568 |
| WoE_Consensus | 0.799 | 0.791 | 0.478 | 0.488 | 0.848 | 0.449 |
In compliance with the OECD principles, the three models are associated with a specific applicability domain (AD) to identify chemicals that fall within the chemical space of the model:
- for M1_KNN a molecule is considered outside the AD if its distance, from the closest neighbor of the training set, is greater than three times the average distance of the training molecules
- for M2_OLS the Leverage method is used (Sahigara et al. 2012)
- a molecule is considered inside the AD of the WoE_Consensus model if it is also inside the AD of both M1_KNN and M2_OLS
The following charts show the predicted (Y) and real (X) values of the models:
| M1_KNN | M2_OLS | Consensus |
|---|---|---|
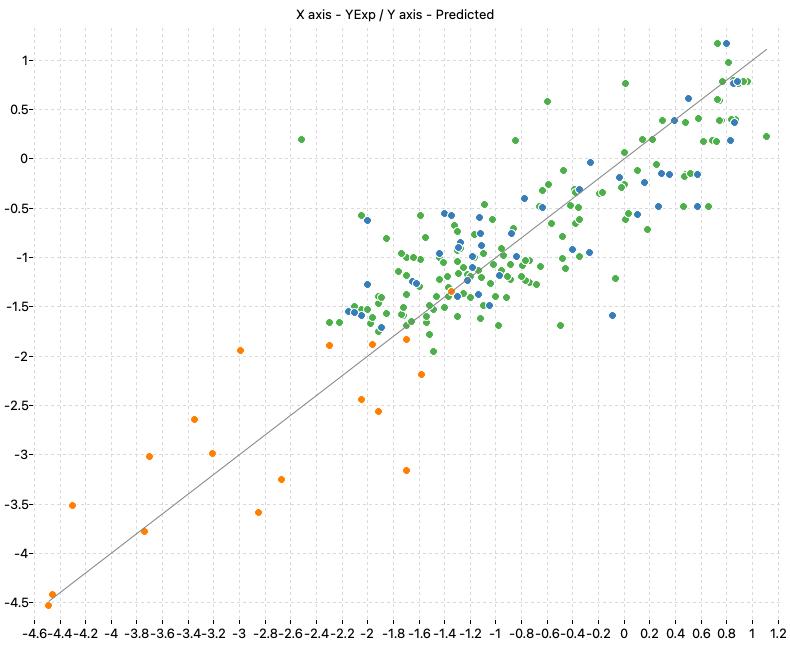 |
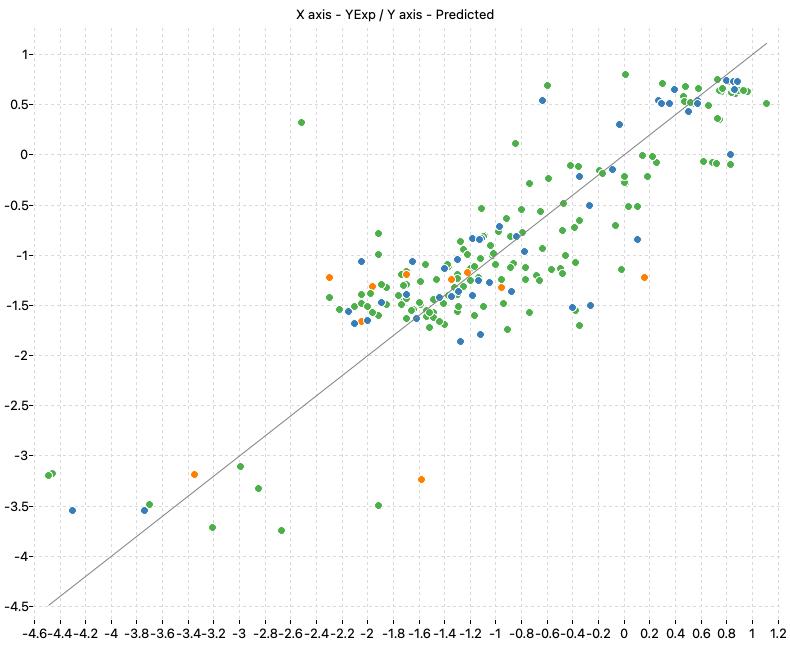 |
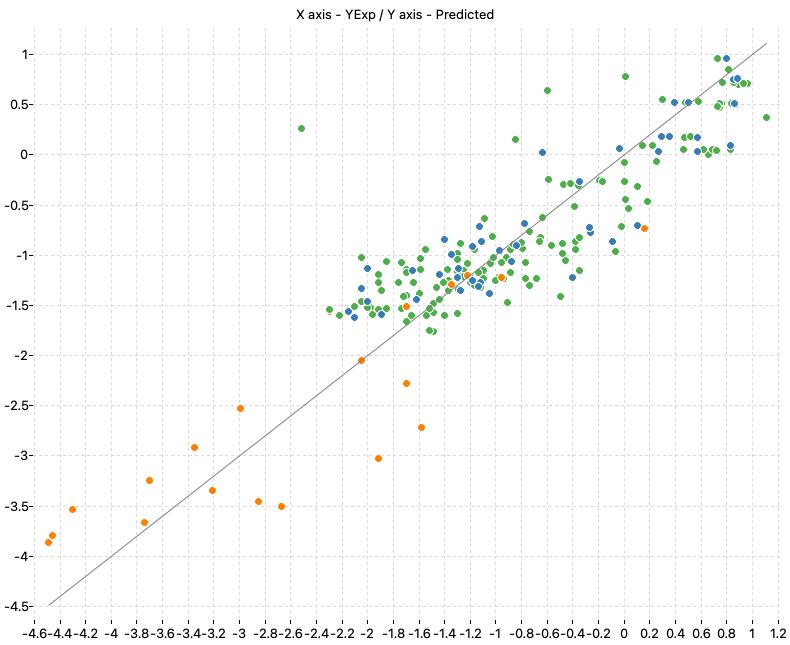 |
PetroRisk
PetroRisk is a tool developed by Concawe which “calculates the environmental exposures and risks resulting from the different lifecycle stages of multi-constituent hydrocarbon substances, using the principles provided by the European Chemical Agency (ECHA) under the EU REACH regulation”.
Starting from version 8.01, PetroRisk makes use of Biomagnification Factor values calculated by using alvaRunner and the BMF project. The PetroRisk manual contains a comparison between predicted BMFs and available experimentally derived BMFs which indicates the model performs well for petroleum hydrocarbons:
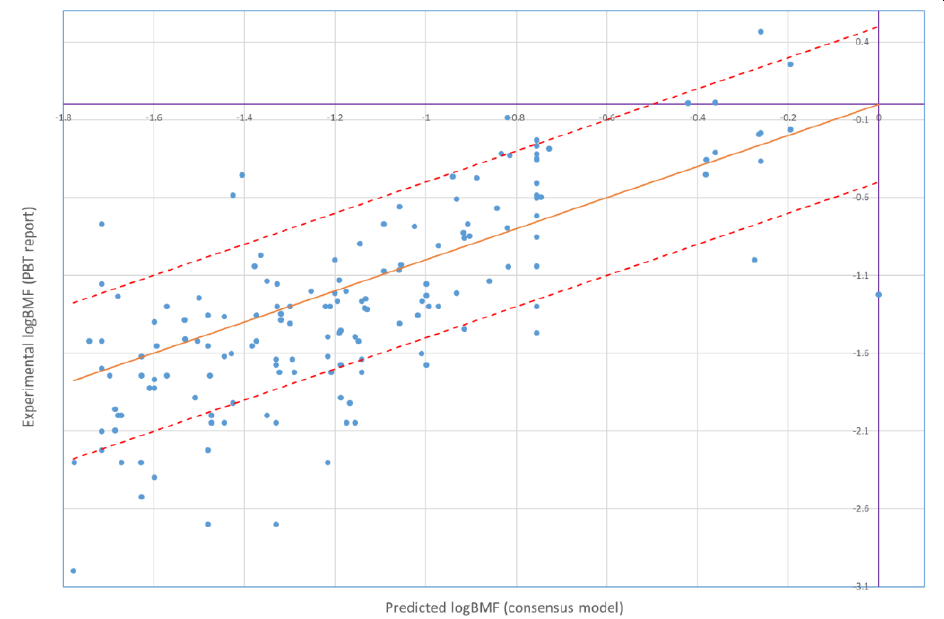
The orange line is the 1 to 1 line (experimental = predicted). The red lines indicate ± 0.5 log units of error from the prediction.
119 out of 158 predictions (or 75%) were less than 0.5 log unit different than the experimental results. 8 (or 5%) of the predictions underestimated the experimental results by more than a 0.5 log unit of error.